What's New
Home Page
My Self
My Family
My Friends
My Work
My Teachers
My Philosophy
My Wife
Favorite Links
Your Comments Please!
My Pictures
Marriage Album
Whats New
What is Today's Topic?
Here I could tell visitors about few important areas of Biotechnology research. These topics will be updated weekly, so that you can see new topics every Monday.
DNA shuffling of a family of genes from diverse species accelerates directed evolution
DNA shuffling is a powerful process for directed evolution, which generates diversity by recombination1,2, combining useful mutations from individual genes. Libraries of chimaeric genes can be generated by random fragmentation of a pool of related genes, followed by reassembly of the fragments in a self-priming polymerase reaction. Template switching causes crossovers in areas of sequence homology. Our previous studies used single genes and random point mutations as the source of diversity3-6. An alternative source of diversity is naturally occurring homologous genes, which provide 'functional diversity'. To evaluate whether natural diversity could accelerate the evolution process, we compared the efficiency of obtaining moxalactamase activity from four cephalosporinase genes evolved separately with that from a mixed pool of the four genes. A single cycle of shuffling yielded eightfold improvements from the four separately evolved genes, versus a 270- to 540-fold improvement from the four genes shuffled together, a 50-fold increase per cycle of shuffling. The best clone contained eight segments from three of the four genes as well as 33 amino-acid point mutations. Molecular breeding by shuffling can efficiently mix sequences from different species, unlike traditional breeding techniques. The power of family shuffling may arise from sparse sampling of a larger portion of sequence space.
Reiterative cycles of shuffling followed by screening or selection has proved to be a useful approach for the evolution of single gene products with enhanced activity3, altered substrate specificity4 or improved protein folding5 and of entire operons with improved function6. When a single starting sequence is used, diversity originates as random point mutations resulting from the polymerase reaction1. Because most point mutations are deleterious or neutral7, the random point mutation rate must be low8 and the accumulation of beneficial mutations and the evolution of a desired function is relatively slow in such experiments. For example, the evolution of a fucosidase from a galactosidase required five rounds of shuffling and screening before a >10-fold improvement in activity was detected4. Naturally occurring homologous sequences are pre-enriched for 'functional diversity' because deleterious variants have been selected against over billions of years of evolution. We therefore wanted to determine whether shuffling of gene families would accelerate the evolution process.
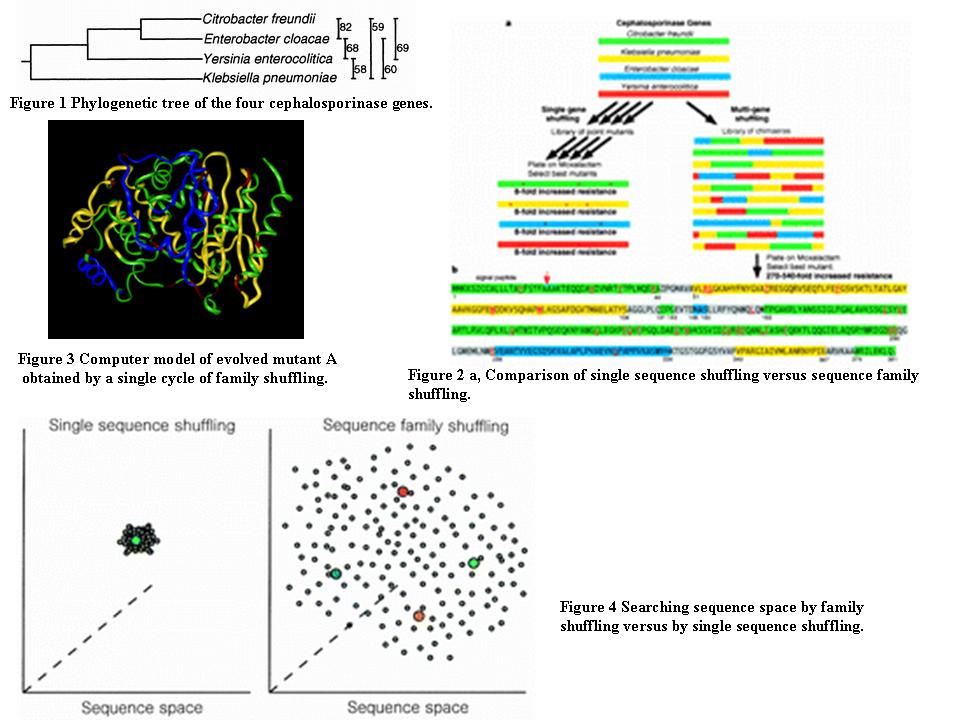
Four 1.6 kilobase (kb) genes encoding class C cephalosporinases, 58�82% identical at the DNA sequence level (Fig. 1), were chosen from four microbial species (Citrobacter freundii9, Enterobacter cloacae10, Klebsiella pneumoniae11 and Yersinia enterocolitica12) and constructed by gene synthesis13 using the original DNA sequence. The four genes were shuffled either individually or as a pool using methods previously described1,3 (Fig. 2a). Equal numbers of Escherichia coli transformants (about 5times 104) expressing the resultant libraries were then plated on media containing a range of concentrations of the antibiotic moxalactam (0.016�32 �g ml-1). After this single round of shuffling, the colonies showing the highest level of resistance to moxalactam were identified and further characterized.
Clones originating from the four single gene libraries showed up to eightfold increases in moxalactam resistance as compared to those expressing the wild-type genes (0.38 to 3.0 �g ml-1 for the Klebsiella and Yersinia genes and 0.75 to 6.0 �g ml-1 for the Citrobacter and Enterobacter genes). In contrast, the best clone (A) originating from the gene family library showed a 540-fold increase in resistance (0.38 to 200 �g ml-1) compared to the wild-type Klebsiella and Yersinia genes and a 270-fold increase (0.75 to 200 �g ml-1) compared to the wild-type Enterobacter and Citrobacter genes (Fig. 2a). Thus 'family shuffling' accelerated the rate of functional enzyme improvement 34- to 68-fold in a single cycle.
A second round of shuffling was performed using the pool of colonies selected in the first round. Plating of about 5times 104 colonies on a range of concentrations of moxalactam yielded three clones which had a further 3.5-fold increased moxalactam resistance over the most resistant clone for the first cycle (clone A). The reduction in the rate of improvement is expected given the limited dynamic range of the bioassay. Based on our experience with other genes3-6, a second round of intra-species shuffling of the selected pools containing the eightfold improved genes would clearly not have achieved the degree of improvement we obtained by two rounds of inter-species shuffling.
The two most resistant clones (A and B) obtained in the first round of family shuffling also showed increased resistance against other beta-lactam antibiotics. Clone A was resistant to cefoxitine, carbenicillin and cephafloridine at 100 �g ml-1each, an improvement over the four wild-type enzymes of 4�16-fold. This was unexpected because in previous results for single gene shuffling of other enzymes the activity for the original substrate was decreased after evolution for increased activity on a new substrate3,4. Plasmid transfer experiments into E. coli NM522 showed that all of the increased resistance was conferred by the plasmid. Sodium dodecyl sulphate�polyacrylamide gel electrophoresis (SDS�PAGE) analysis of periplasmic extracts showed that the expression level of the four wild-type cephalosporinases was indistinguishable from that of the mutant clones A and B (data not shown). Antibiotic bioassays showed that all of the moxalactam was degraded by cells expressing chimaeric enzyme A or B, but not by cells expressing any of the four wild-type enzymes. This indicates that the specific activity of the chimaeric moxalactamases was improved. However, our attempts to obtain and compare the kinetic profiles of the wild-type and chimaeric enzymes failed because of the very low activity of the wild-type enzymes.
The genes encoding the new moxalactamases from these two clones were sequenced. Clones A and B were both found to be chimaeras of the genes from Citrobacter, Enterobacter and Klebsiella. Both clones had a similar overall structure containing eight segments resulting from seven crossovers. Because of local DNA homology the crossover location could not be defined more exactly than indicated by the grey segments shown in Fig. 2b. The crossovers occurred in areas where the two genes had 14�37 base pairs (bp) of nearly identical sequence (average 57% GC). In addition, chimaera A had 47 DNA point mutations, 6 of which were silent, resulting in 33 amino-acid substitutions scattered throughout the gene that did not exist in any of the four parental enzymes. Chimaera B had 14 amino-acid substitutions, of which 12 were identical to those in chimaera A.
A model of the best chimaeric moxalactamase, chimaera A, was created from the known structure of the Enterobacter cloacae AmpC enzyme14 (Fig. 3). Although 37% of the amino acids (142 residues) of the chimaeric clone A differ from the Enterobacter cloacae enzyme for which the crystal structure is known, after energy minimization the predicted structure of the alpha-chain backbone of the A chimaera remained nearly identical to the known structure (r.m.d.s. deviation of 0.766 ?). This model shows that two crossovers occurred in loop or random coil regions separating alpha-helical and beta-sheet structures, and one occurred inside the C-terminal alpha-helix. The remaining five crossovers could have occurred in loops, alpha-helices or beta-sheets; therefore it is unclear whether the crossovers preferentially occurred in loops separating structural elements. The conserved catalytic residues S64, K67, Y150 and K315 of the Enterobacter enzyme14 were retained.
Although shuffling of a single gene creates a library of genes that differ by only a few point mutations1-6, the block-exchange nature of family shuffling creates chimaeras that differ in many positions. For example, in previous work a single beta-lactamase gene was shuffled for three cycles, yielding only four amino-acid mutations3, whereas a single cycle of family shuffling of the four cephalosporinases resulted in a mutant enzyme which differs by 102 amino acids from the Citrobacter enzyme, by 142 amino acids from the Enterobacter enzyme, by 181 amino acid from the Klebsiella enzyme and by 196 amino acids from the Yersinia enzyme. The increased sequence diversity of the library members obtained by family shuffling results in a 'sparse sampling' of a much greater portion of sequence space15, the theoretical collection of all possible sequences of equal length, ordered by similarity (Fig. 4). Selection from 'sparse libraries' allows rapid identification of the most promising areas within an extended sequence landscape (a multidimensional graph of sequence space versus function)15. However, the sparseness also decreases the likelihood of immediate location of the area's best sequence, the local optimum. Subsequent shuffling of the selected sequences allows further exploration of the still vast intermittent sequence space at an increased sampling density. Although the search algorithm remains unchanged, the scale of the searched area decreases with each cycle until no further improvement occurs.
Because the species definition in eukaryotes is based on the ability to exchange genetic information, the family shuffling that occurs in natural evolution and in classical breeding is generally restricted to the diversity existing within a single species. DNA shuffling can successfully mix genes from diverse species because single genes can tolerate much higher mutation densities16 than whole genomes. The high degree of DNA homology that is required for traditional breeding of plants and animals is therefore not required for the molecular breeding of single genes and gene clusters.
Methods
Gene synthesis. All four 1.6 kb genes were separately assembled from 60-mer synthetic oligonucleotides in a single assembly reaction13. These genes were cloned into pBR322 downstream of the beta-lactamase promoter and in place of the native beta-lactamase gene. The DNA sequence and drug resistance of each construct was confirmed.
Library construction. The presence of chimaeric genes in the library made by family shuffling was confirmed by restriction fragment length polymorphism (RFLP) analysis of individual clones. Each of the eight clones analysed had an RFLP pattern distinct from each other as well as from those of the parental sequences. The library thus contained a diverse array of chimaeric sequences. The second cycle of shuffling was performed on the pool of chimaeras selected in the first round.
Drug resistance. An equal number of transformants were plated on medium containing moxalactam. The numbers were based on the number of tetracyclin-resistant colonies after transformation. The resistance of the selected clones to a variety of beta-lactam antibiotics was measured in 96-well plates in which each antibiotic was serially diluted (1:1) into culture medium containing the test organism. Assays for all native and mutant clones were done in quadruplicate.
Moxalactamase assays. The moxalactam degradation activity of whole cells was measured by incubating an overnight culture of bacteria with 34 �g ml-1of moxalactam in LB media. After incubation for 16 h the concentration of moxalactam in the sterilized supernatant was measured by bioassay on plates inoculated with E. coli NM522, using filter discs saturated with serial twofold dilutions of the supernatants. A moxalactam standard curve was used to determine the fraction of moxolactam degraded by the host cell alone, the four wild-type constructs and the two chimaeric mutants.
Modelling. The deduced amino-acid sequence of chimaeric mutant A was aligned with the wild-type Enterobacter sequence using HOMOLOGY (Biosym, San Diego) and the calculated coordinates were assigned to the chimaeric protein. The modelled structure was constructed using a three-step protocol that involved an energy minimization to relieve steric hindrance, molecular dynamics calculations to find the lowest energy conformation followed by another energy minimization to provide the final structure.
Received 23 June 1997;accepted 8 October 1997
------------------
References
1. Stemmer, W. P. C. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl Acad. Sci. USA 91, 10747-10751 (1994). | PubMed | ISI |
2. Stemmer, W. P. C. Searching sequence space. Bio/Technology 13, 549-553 (1995). | ISI |
3. Stemmer, W. P. C. Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389-391 (1994). | PubMed | ISI |
4. Zhang, J., Dawes, G. & Stemmer, W. P. C. Evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc. Natl Acac. Sci. USA 94, 4504-4509 (1997). | ISI |
5. Crameri, A., Whitehorn, E., Tate, E. & Stemmer, W. P. C. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nature Biotech. 14, 315-319 (1996). | PubMed | ISI |
6. Crameri, A., Dawes, G., Rodriguez, E., Silver, S. & Stemmer, W. P. C. Molecular Evolution of an arsenate detoxification pathway by DNA shuffling. Nature Biotech. 15, 436-438 (1997). | PubMed | ISI |
7. Moore, J. C. & Arnold, F. H. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nature Biotech. 14, 458-467 (1996). | PubMed | ISI |
8. Zhao, H. & Arnold, F. H. Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res. 25, 1307-1308 (1997). | Article | PubMed | ISI |
9. Lindberg, F. & Normark, S. Sequence of the Citrobacter freundii OS60 chromosomal ampC beta-lactamase gene. Eur. J. Biochem. 156, 441-445 (1986). | PubMed | ISI |
10. Galleni, M. et al. Sequence and comparative analysis of three Enterobacter cloacae ampC beta-lactamase genes and their products. Biochem. J. 250, 753-760 (1988). | PubMed | ISI |
11. Leiza, M. G. et al. Gene sequence and biochemical characterization of FOX-1 from Klebsiella pneumoniae, a new AmpC-type plasmid-mediated beta-lactamase with two molecular variants. Antimicrob. Agents Chemother. 38, 2150-2157 (1994). | PubMed | ISI |
12. Seoane, A., Francia, M. V. & Garcia Lobo, J. M. Nucleotide sequence of the ampC-ampR region from the chromosome of Yersinia enterocolitica. Antimicrob. Agents Chemother. 36, 1049-1052 (1992). | PubMed | ISI |
13. Stemmer, W. P. C., Crameri, A., Ha, K. D., Brennan, T. M. & Heyneker, H. L. Single-step PCR assembly of a gene and a whole plasmid from large numbers of oligonucleotides. Gene 164, 49-53 (1995). | Article | PubMed | ISI |
14. Lobkovsky, E. et al. Evolution of enzyme activity: crystallographic structure at 2 ? resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc. Natl Acad. Sci. USA 90, 11257-11261 (1993). | PubMed | ISI |
15. Kauffman, S. The Origins of Order (Oxford University Press, Oxford, (1993)).
16. Eigen, M. Steps Towards Life: a Perspective on Evolution (Oxford University Press, Oxford, (1992)).